 Figure 1
Figure 1
Figure 1
Figure 1
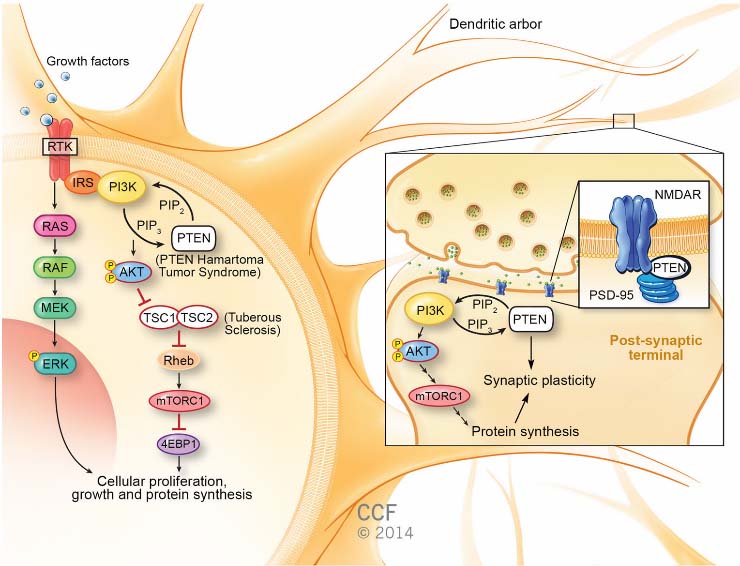
Germline mutations in PTEN, which encodes a widely expressed phosphatase, was mapped to 10q23 and identified as the susceptibility gene for Cowden syndrome, characterized by macrocephaly and high risks of breast, thyroid, and other cancers. The phenotypic spectrum of PTEN mutations expanded to include autism with macrocephaly only 10 years ago. Neurological studies of patients with PTEN-associated autism spectrum disorder (ASD) show increases in cortical white matter and a distinctive cognitive profile, including delayed language development with poor working memory and processing speed. Once a germline PTEN mutation is found, and a diagnosis of phosphatase and tensin homolog (PTEN) hamartoma tumor syndrome made, the clinical outlook broadens to include higher lifetime risks for multiple cancers, beginning in childhood with thyroid cancer. First described as a tumor suppressor, PTEN is a major negative regulator of the phosphatidylinositol 3-kinase/protein kinase B/mammalian target of rapamycin (mTOR) signaling pathway—controlling growth, protein synthesis, and proliferation. This canonical function combines with less well-understood mechanisms to influence synaptic plasticity and neuronal cytoarchitecture. Several excellent mouse models of Pten loss or dysfunction link these neural functions to autism-like behavioral abnormalities, such as altered sociability, repetitive behaviors, and phenotypes like anxiety that are often associated with ASD in humans. These models also show the promise of mTOR inhibitors as therapeutic agents capable of reversing phenotypes ranging from overgrowth to low social behavior. Based on these findings, therapeutic options for patients with PTEN hamartoma tumor syndrome and ASD are coming into view, even as new discoveries in PTEN biology add complexity to our understanding of this master regulator.